

Gaussian16windows版是软件的电脑PC版本,这是一款模拟建模软件,在软件中用户可以进行分子元素的模拟,你可以将分子的形态在电脑中完全模拟出来,在电脑上轻松完成分子的结合,方便用户根据分子的结构,进行分子特性的探索,软件适用于各大领域,特别是科学家、生物学家等等,通过使用这款软件,可以让实验变得更加简单。从量子力学的基本定律出发,在各种不同的化学环境中预测分子结构、能量、振动频率、分子性质与反应。Gaussian 16的模型既可以应用于稳定的体系与化合物,也适用于实验中很难或不可能观察到的体系或化合物(例如,生存周期很短的中间体和过渡态结构)。
版本特色
1、支持在限制性优化、柔性扫描等任务中,使用新的广义内坐标定义方式
2、通过 geom=GIC 声明一个新的输入段落编写下列内容
可以定义几何变量名,如 HOH=A(2 , 1 , 3);对变量进行运算;运算中修改数值;使用条件控制语句;协同的限制性优化、柔性扫描
4、支持的几何变量函数:
原来的键长、键角、二面角、线性弯曲 R、A、D、L;
原子坐标分量 X、Y、Z;
n个原子的重心分量 XCntr、YCntr、ZCntr (如XCntr(1 , 12 - 15 , 27) 表示这5个原子重心的原子坐标 X 分量)
两向量的点积 DotDiff(i , j , k , l) = ( i - j ) · ( k - l ),其中 i , j , k , l 为原子序号对应的坐标向量
5、支持的数学运算:加、减、乘、除、乘方,开方 SQRT,指数 EXP,三角函数 SIN、COS、TAN,ARCCOS
6、支持的操作:
冻结 Frozen;
柔性扫描 StepSize=x, NSteps=n;
(恢复)指定初值 Value=X;
增加 / 不增加 / 删除冗余内坐标 Active / Inactive / Kill;
输出值 PrintOnly;
指定初始Hessian中的相应对角项数值 ForceConstant = x;
在特定数值区间内时激活选项 Min, Max;
条件控制语句 OnlyIf, IfNot
使用示例:
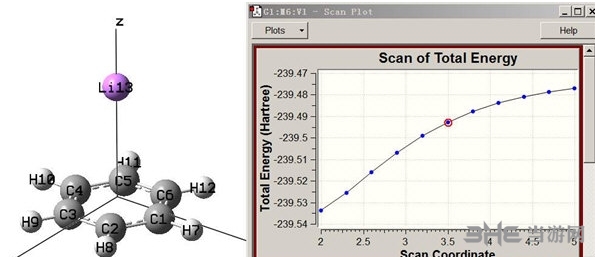
7、计算两分子/两分子片段间的电子能量转移速率(rate of EET),可用于考察光化学中的常见过程如 M* + A --> M + A*
8、新方法:
半经验:PM7;
泛函:M08系列(?),MN15,MN15L;
双杂化:DSDPBEP86,PBE0DH,PBEQIDH
9、更好的 GEDIIS 几何优化方法
10、提供 opt=recalc 选项代替原来隐藏的 IOp(1/40=n) 选项,在优化过程中每隔n步计算一次精确 Hessian
11、TD-DFT 解析 Hessian (可用于激发态频率、红外拉曼、过渡态优化、IRC);(注:不支持TD+SCRF的解析频率,即只能不加溶剂做)
12、提供高斯与 C++, Fortran, Python, Perl 的接口
13、优化核数较多时高斯的并行效率
14、可以指定 Gaussian 使用的 CPU 线程号(其实这个功能完全可以通过排队程序管理):
使用 %CPU=0‑7 可以指定 Gaussian 使用第1个至第8个线程,在只有8个线程的机器上等效于 %nprocshared=8
使用 %CPU=0-16/2 指定 Gaussian 使用第 1、3、5、7、9、11、13、15 号线程,主要用于防止挤在某核超线程出来的两个线程中、及在多 CPU 的机器上均衡负载
P.S. Gaussian 官方推荐关掉超线程了,WTF!抽空发个评测打脸。
15、支持在命令行或环境变量中定义原来的各 %link0 选项
16、修改了一些默认设置
全面提升各种默认积分精度:
默认积分精度从 1E-10 修改为了 1E-12,等价于默认 int=ACC2E=12
在所有 DFT 计算中默认 int=UltraFine (吃饱了撑的?)
CPHF 默认 int=SG1
修改默认内存使用量至 %mem=800MB
SCRF默认算法修改为“symmetric form of IEFPCM”,之前没有这个功能
软件功能
1、分析红外和 静态和动态拉曼强度(高频& DFT;MP2 IR)。
2、振动频率和正常模式, 包括显示输出限制指定原子/残留/模式(可选模式排序)。
3、可重新开始的分析高频和DFT频率。
4、莫:MM ONIOM频率包括电子嵌入。
5、Pre-resonance拉曼光谱(高频和DFT)。
更新内容
1、支持TD频率、TS优化与IRC计算
2、非简谐振动光谱
3、电子振动光谱等
4、EOMCC优化
5、支持GPU计算提高性能
6、支持Fortran、C、Perl与Python的接口














-
本类热门推荐本类热门标签
-
详情
 Why数学图像生成工具 绿色版v3.0
2.07MB / 3分
Why数学图像生成工具 绿色版v3.0
2.07MB / 3分
-
详情
 金山打字通 绿色版V2.2.0.55
24.9MB / 3分
金山打字通 绿色版V2.2.0.55
24.9MB / 3分
-
详情
 亿图公式编辑器EdrawMath 官方版v1.0
48.9MB / 3分
亿图公式编辑器EdrawMath 官方版v1.0
48.9MB / 3分
-
详情
 MathMarkEdit (数学公式编辑器)官方版v1.0
4.52MB / 3分
MathMarkEdit (数学公式编辑器)官方版v1.0
4.52MB / 3分
-
详情
 algodoo物理沙盒 官方最新版v2.0.0
35.87MB / 3分
algodoo物理沙盒 官方最新版v2.0.0
35.87MB / 3分
-
详情
 WPS公式编辑器 官方最新版v3.0
9.78MB / 3分
WPS公式编辑器 官方最新版v3.0
9.78MB / 3分
-
详情
 ChemOffice2018 官方正式版v18.1.0
371.55MB / 3分
ChemOffice2018 官方正式版v18.1.0
371.55MB / 3分
-
详情










-
2 1stOpt软件







装机必备软件
网友评论